
1. INTRODUCTION
Type 2 diabetes cases are increasing worldwide, but it is promising to handle the number by exercising and diet control.1 The study proved that hypoglycaemic drugs that belong to the Meglitinide class of short-acting insulin secretagogues could constrain blood glucose by accelerating the insulin release from the pancreas, which is ATP-supported potassium channels β cells.2 The deadly illness needs regulation, and it could possibly cause lifelong difficulties like peripheral vascular disease due to probable disorders in blood circulation, stroke, which minimises blood flow towards the brain, blindness, cardiovascular disease, diabetic neuropathy that damages the nerve and renal failure because of kidney damage.3–5 Diabetes is also considered to be a protein and carbohydrate reduction with the metabolism of fat due to a deficiency of insulin that causes hyperglycemia.6
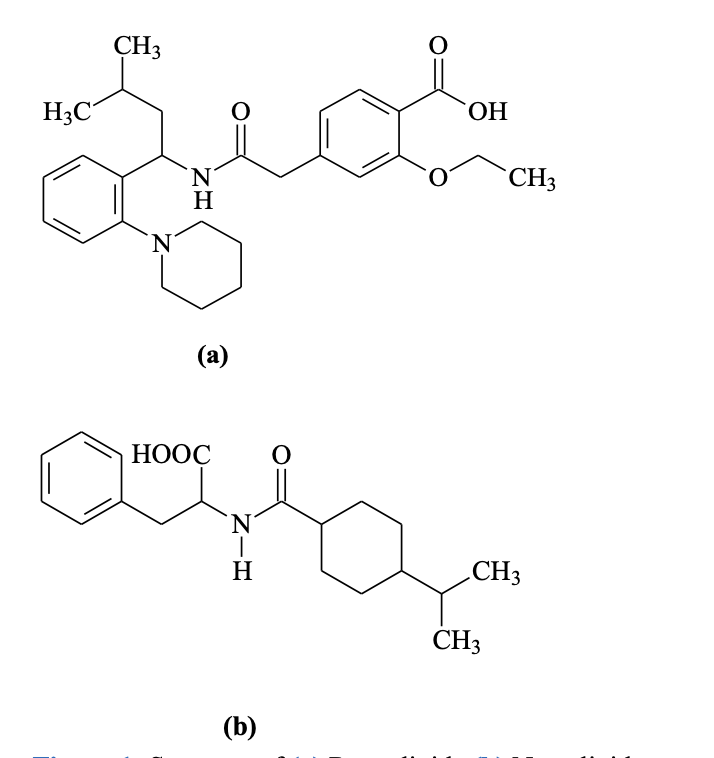
The meglitinide agent, repaglinide (Figure 1a), is used for antidiabetic management of type 2 diabetes mellitus. It is chemically known as S(+)2-ethoxy-4(2((3- methyl-1-(2-(1-piperidinyl) phenyl)-butyl) amino)-2-oxoethyl) benzoic acid. The other D-phenylalanine derivative, nateglinide (Figure 1b), also has been approved for treating diabetes mellitus. So, it is necessary to understand the drug development process, which is essential to support scientific and commercial success. Ensuring an active pharmaceutical ingredient (API) ‘s physical and chemical assets and pharmaceutical formulation is vital for regulatory agreement and therapeutic accomplishment. The major challenge for drug development is manufacturing and controlling components, ensuring that the compound and product’s chemical and physical properties are examined at all crucial phases of the pathway. Stability studies are necessary to verify and provide the identity, purity and potency of ingredients and formulated products. The analytical method’s limitations included controlled applicability to individual problems. These methods can be challenging to apply for complex issues and time-consuming, labour-intensive processes. Their applicability is also limited in real-world problems and difficulties when combined with external factors. These limitations make methods less versatile but provide adaptability to the approximate methods that could routinely solve complicated issues in various fields. As a result, analytical methods’ limitations can indicate increased reliance on approach methods, which could help the researchers understand the method’s applicability and select a suitable technique for the analysis. Due to its therapeutic importance, several methods, such as ultraviolet-visible spectrophotometry (UV–Vis), high-performance liquid chromatography (HPLC), high-performance thin-layer chromatography (HPTLC), ultra–pressure liquid chromatography (UPLC), high-performance liquid chromatography-mass spectrophotometry (HPLC-MS) and ultra–pressure liquid chromatography-mass spectrophotometry (UPLC-MS) are based on different techniques had been reported in API, pharmaceutical formulations and biological fluids. The main aim of antidiabetic drugs is to serve humans and free them from promising illness and disease prevention. For medicines like NTG and RPG to provide their projected target,7,8 they would be protected from contamination or other obstacles that harm humans.

The current review focuses on determining and quantifying NTG and RPG in API, pharmaceutical formulations and biological fluids. It also signified the introduction of analytical techniques and their advantages and limitations in quantifying the drugs. It also emphasizes advancing the reported practice from the older UV-Vis approach to innovative hyphenated methods. It might help new researchers from academia and industry to understand their functions and capabilities, which could open an option whether these methods are still the best or are being supplanted with more advanced techniques.
2. ANALYTICAL TECHNIQUES
2.1. SPECTROPHOTOMETRY
UV–Vis spectrophotometry is a valuable and easy technique for a drug’s qualitative and quantitative determination due to its simple, low-cost technique, without pretreatment prior to analysis (Scheme 1). It also required less time and labour intake. This technique, mainly used in pharmaceutical formulation analysis, has increased based on the redox and complexation reaction.

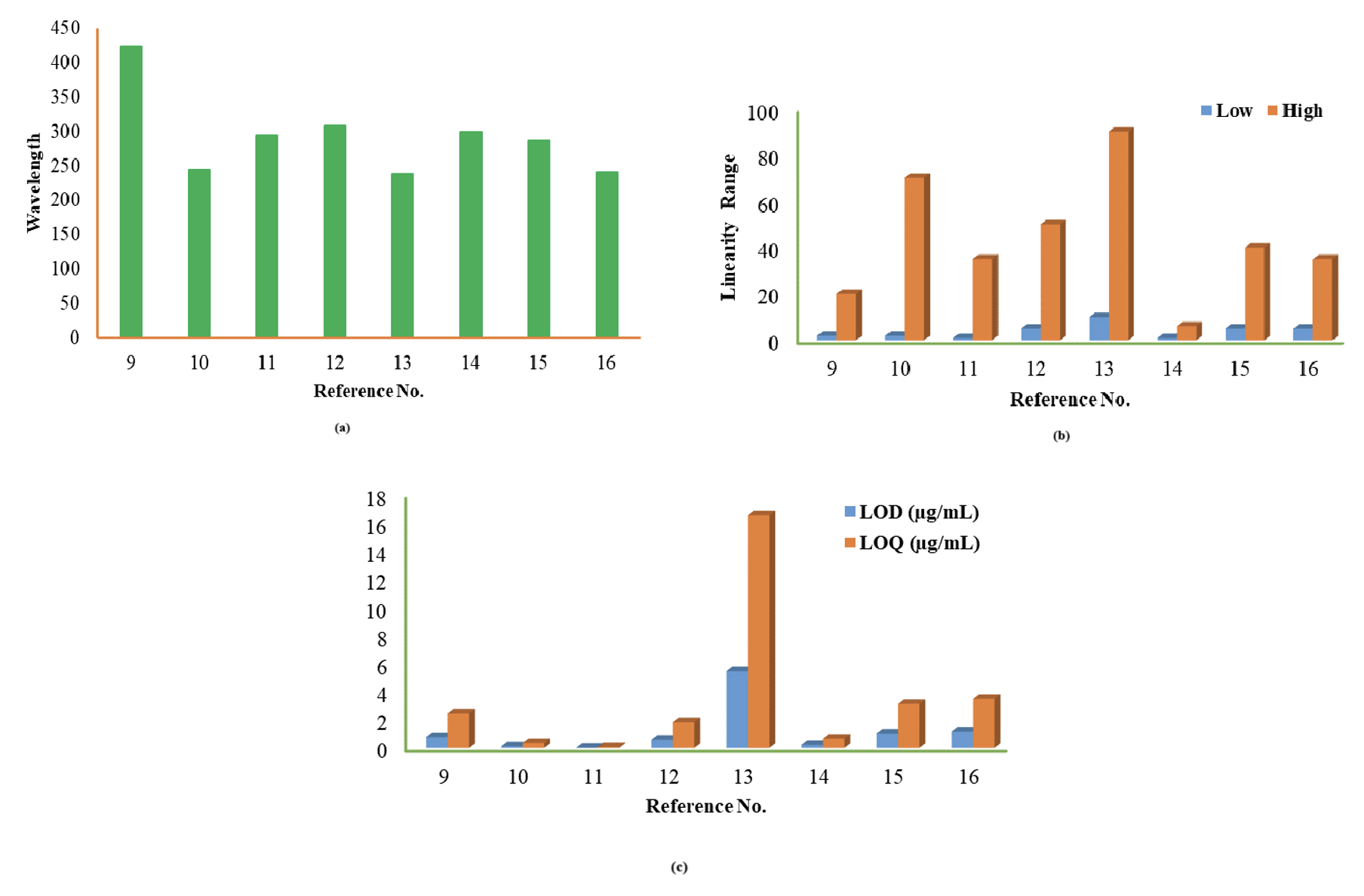
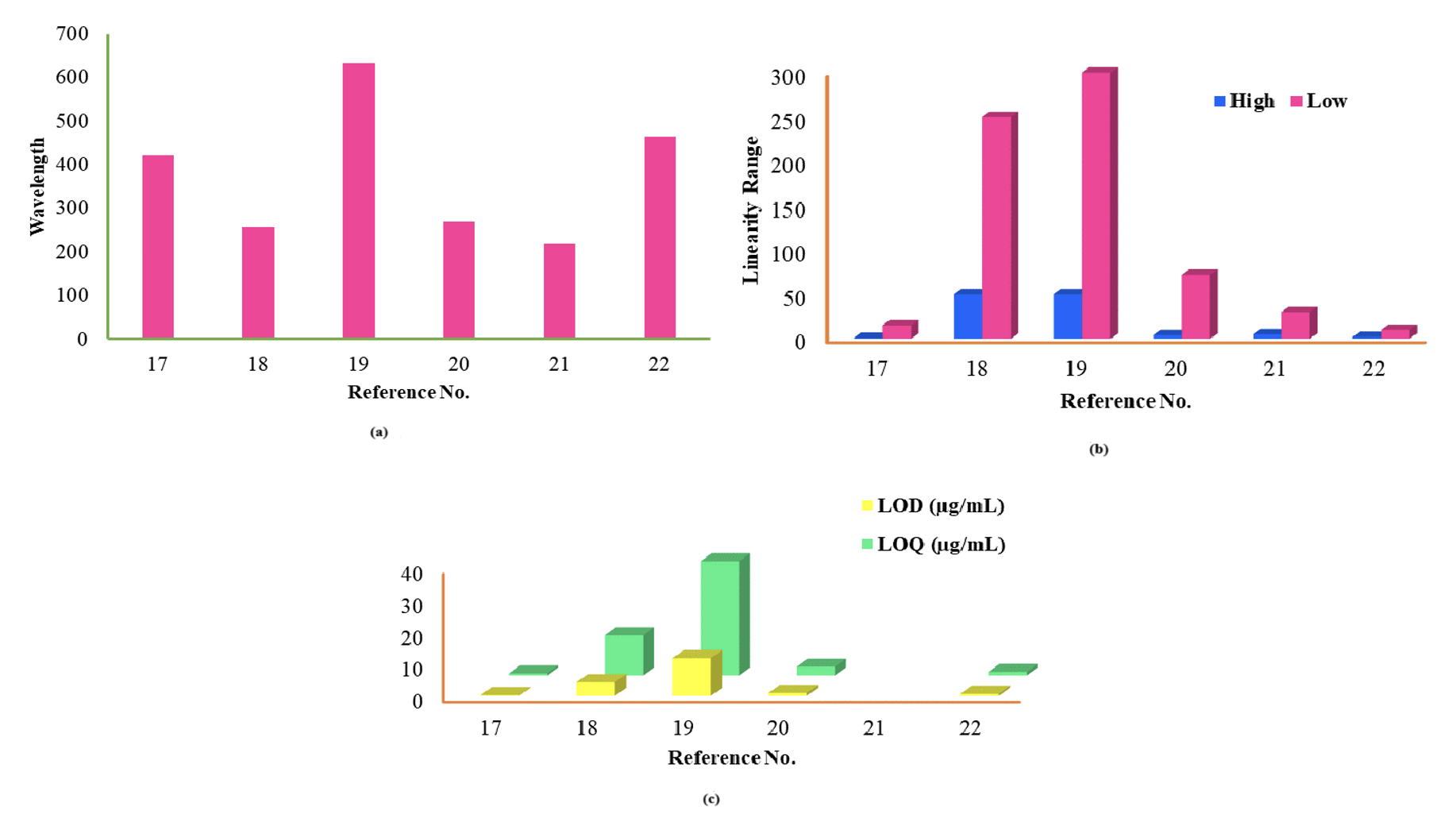
The detailed investigation based on the specificity, sensitivity, and broader range of each reported procedure were included in Table 1,9–22 and studies elaborate on all reported procedures for both. Nearly all the analytical determinations used methanol as a diluent for RPG quantification11–13,15,16 at different wavelengths. However, the procedure showed excellent linearity in bulk and pharmaceutical formulations. The precision results were within the acceptable limit. The developed method9 is based on the kinetic spectrophotometry applied response surface methodology combined Box–Behnken design, which reduced the uses of reagents and solvents with the minimum number of trials and supported the eco-friendly green analysis. The % assay of the RPG in tablets was entirely satisfactory, about 98.24% as per the Indian Pharmacopoeia.10 The degradation results also confirmed its value within the permissible range. The % recovery was 97.92–101.35%, and the % RSD was less than 2% (Figure 2). The literature continued to reveal the NTG quantification17–22 with a wider dynamic range in bulk and pharmaceutical formulations (Figure 3). The determination was based on visible spectrophotometry.17,19,22 It showed no interference from excipients due to higher wavelength in the visible region with an acceptable % recovery. However, minor positive interference was detected in the occurrence of a significant amount of excess excipients such as glucose, lactose, Arabic gum, sodium chloride, sucrose and starch. Few developed procedures have used18,22 methanol as a diluent that focuses on precise results and confirms that all the reported procedures are susceptible to accessing the drug concentration in different matrices.


2.2. HIGH-PERFORMANCE LIQUID CHROMATOGRAPHY (HPLC)
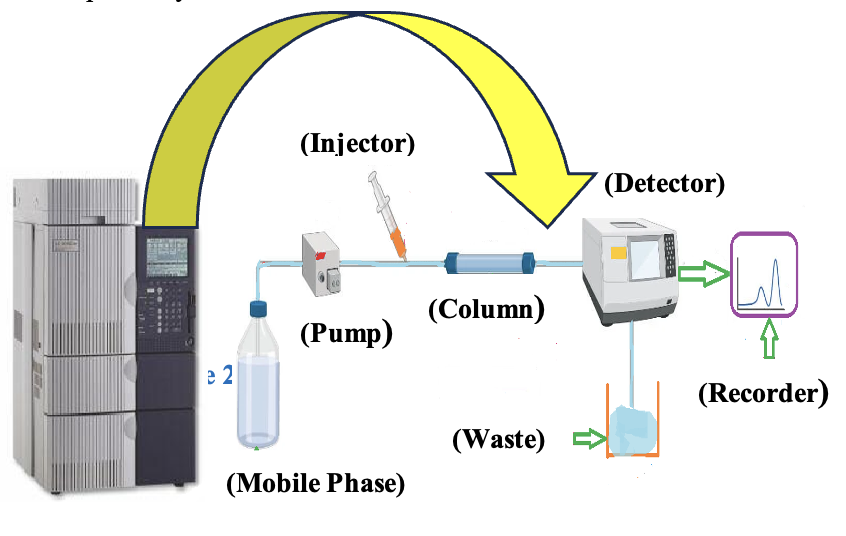
All the chromatography methods, from traditional to hyphenated, are more accurate and precise depending on the available analyte’s characteristics and present stationary and mobile phases. Based on that, different chromatography techniques were established for pharmaceutical analysis. HPLC has recently shown much interest due to its ease of instrumentation, selectivity, and high specificity for drug analysis (Scheme 2). As usual, it is a wholly improved type of column chromatography, in which a solvent is constrained through a column at high pressures rather than allowed to move under gravity. As an outcome, it is significantly faster. Generally, using a non-polar stationary phase for polar analytes is called reverse phase chromatography, whereas for non-polar components, using a polar mobile phase is known as normal phase chromatography.

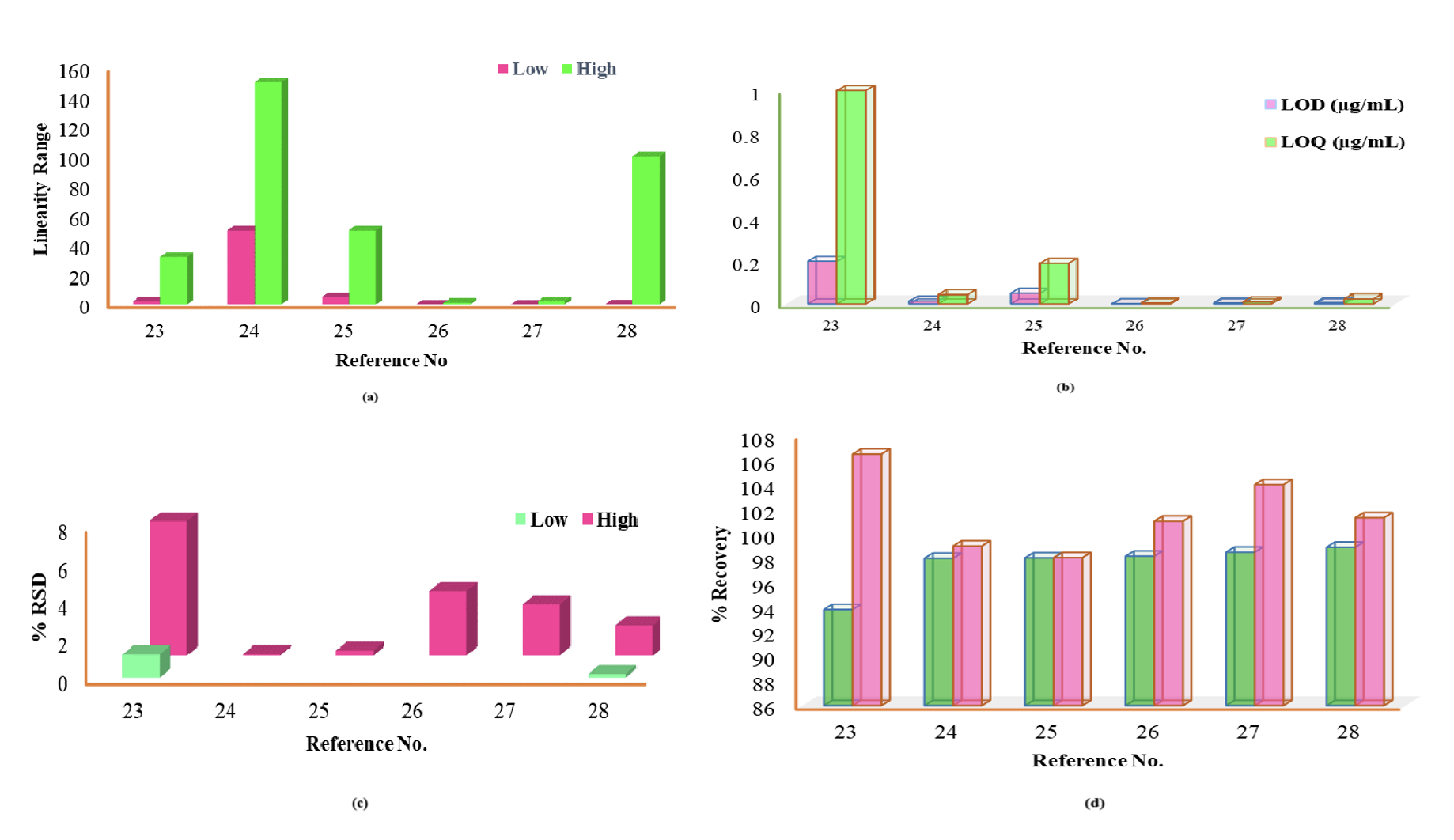
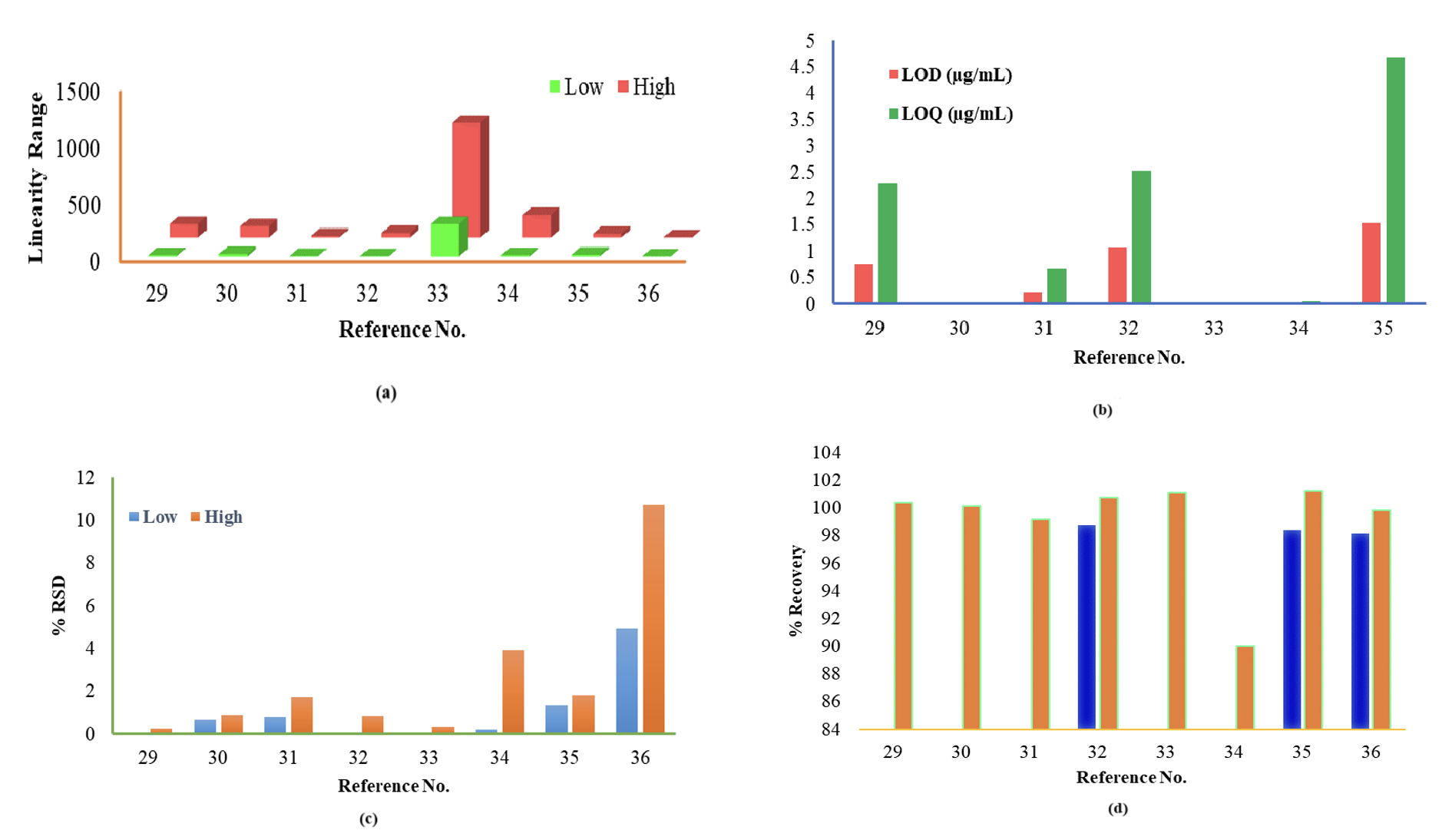
However, it always needs to be kept in mind during method development that solvents and reagents must be used less to be eco-friendly and use a white analytical methodology. The determination of RPG23–28 and NTG29–36 employed varied column types, and a combination of different mobile phases containing buffers and an organic modifier could be used to develop the method for their quantification (Table 2).
The reported method was first applied to determine RPG instantaneously in the presence of remdesivir (RDV) and dexamethasone (DXM) using a lesser organic solvent in the mobile phase.23 It was a prominent procedure to produce more precise results with a large number of quality control samples. The sample recoveries agreed with their respective label claims in the formulation with a lesser retention time,24,25 and formulation excipients did not restrict RPG estimation, demonstrating appropriateness for identifying counterfeit drugs.28
Few analytical procedures are selective and can quantify RPG in biological fluids26,27 with excellent accuracy, precision, and higher therapeutic stability range, including the liquid extraction practice that was sensitive and reproducible with enhanced extraction and development efficiency.26
However, the bioanalytical-based HPLC procedure that mingled with the fluorescence detector required 40ºC as column temperature and a run time of 23 min,27 which also included a simple protein precipitation process for its determination and validation. The precision results were assessed based on the %RSD value and calculated as 8.30% and less.
In comparison, the accuracy was estimated in terms of % recovery within 98.6–112% for the sample concentration in the range of 10–2000 ng/mL in rat plasma, which was considered accurate, precise and reproducible as per the FDA guidelines. The NTG quantification enhanced with Box–Behnken helped to reduce the analysis time and solvent consumption.29 The retention time was decreased with higher reproducibility, increasing the method’s quantification capability.30–35 Waters Nova Pak eluted the NTG within 1.4 minutes at room temperature.35 Parent drug molecule detection could be possible at temperatures over 30ºC and higher run time.36 The graphical representation of the valuable data (Figures 4 and 5) demonstrates its respective quantitative perspective to quantify RPG and NTG, respectively.


2.3. HIGH-PERFORMANCE THIN LAYER CHROMATOGRAPHY (HPTLC)
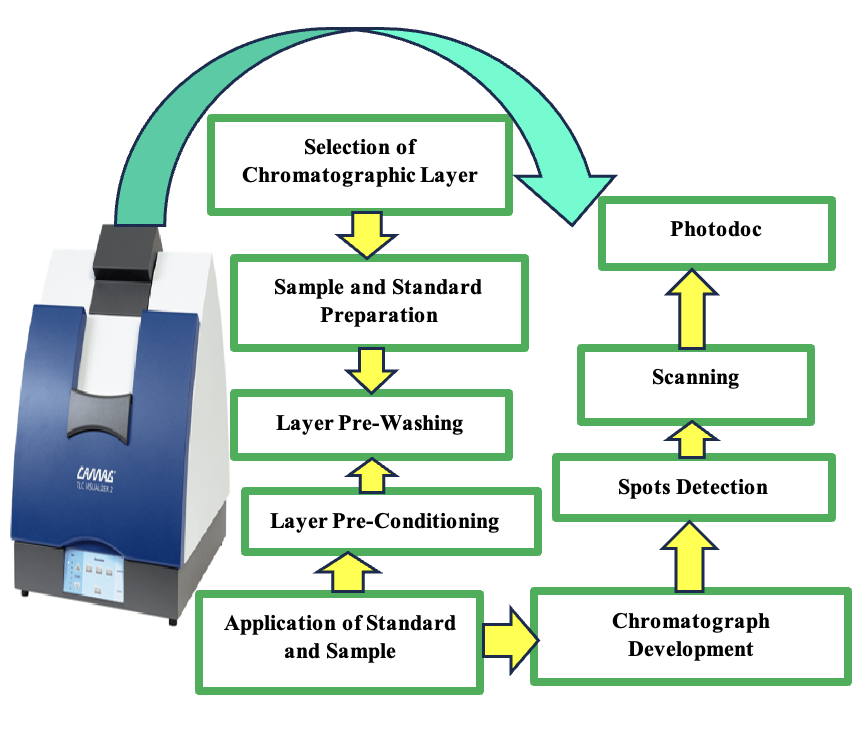
High-performance thin-layer chromatography (HPTLC) is the most convincing and cumulative form of thin-layer chromatography (Scheme 3). It concerns about the chromatographic layers based on different functionalities, and these principles were applied to develop advanced equipment for all phases in the method development. It includes a standardized process based on efficient qualitative and quantitative testing approaches depending on scientific evidence. It also encounters all the quality needs of a current analytical research laboratory. The progress in HPTLC aims to increase the compound’s resolution to be separated and facilitate the substance’s quantitative analysis.

The developments could be with the stationary phase, likely higher-quality chromatography plates that contain significantly thinner particle sizes that permit improved resolution with lower LODs, higher sensitivity, and low cost.37 The reported method was applied to quantify NTG and its present impurities.38 The advantage of the procedure was to use fewer organic solvents, which could be eco-friendly and cost-effective due to reduced methanol consumption with a linearity range of 100–600 µg/band. The procedure comprehensively determines its impurities 1 and 2 in bulk and pharmaceutical formulation with LOD and LOQ 4.17 (ng/band), 0.28 (ng/band), 6.21 (ng/band) and 1.81 (ng/band), respectively. Applying the preparative liquid chromatography method also showed no excipient interference from the formulation during impurity isolation.
The NTG determination could be quantified with other drug combinations like with metformin applying ethyl acetate, chloroform and acetic acid (6:4:0.1, v/v/v) as a mobile phase with pre-coated silica gel (60 F–254) on aluminium plates as a stationary phase which detected within the range of 200–2400 ng/band at 216 nm.39 The procedure had a higher recovery percentage of 99.72, with the potential to quantify simultaneously from formulation without any intervention from the excipients. However, a similar stationary phase could be used to determine RPG from other combinations like metformin and melamine with different mobile phase mixtures of methanol, chloroform and acetic acid (1: 8.5: 0.5, v/v/v), which detected at 240 nm40 within the range of 0.2–1.5 μg/band. Whereas the other solvent system combined with methanol, ammonia, chloroform and acetic acid (1.5: 0.9: 7.5: 0.1, v/v/v/v) could able to determine its purity and related impurities with a wider dynamic range of 50–800 ng/spot. The percentage accuracy showed a higher % recovery between 98.79 and 99.61.41 Therefore, HPTLC methods had many benefits compared to LC, which included one standard plate that could analyse simultaneous samples with reduced equilibrium time and higher mobile phase pH. It could also hold larger samples and consume less solution with decreased run time without solvent pretreatment. The stability-indicating assets indicated that the substances could possibly be assessed in the occurrence of their degradation product. Thus, it can simultaneously evaluate their degradation products in stability samples in the industry.
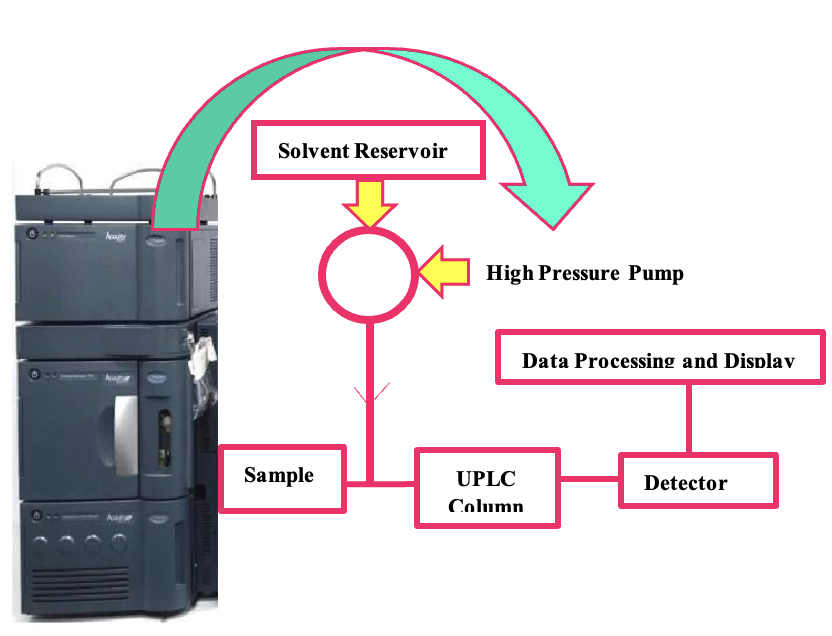
2.4. ULTRA-PRESSURE LIQUID CHROMATOGRAPHY (UPLC)
Ultra-pressure liquid chromatography (UPLC) is a new practice that creates new potential, specifically involving reduced time and solvent utilization. The UPLC chromatographical system uniquely resists the system’s high back pressures by connecting particular types of analytical columns (Scheme 4). In conventional HPLC, the particle size selection should be an agreement in which the smaller the particle size, the higher the influence of increasing the column back pressure, which could be a restraint of employing such columns.

The column diameters of 1 and 2.1 mm could cause similar difficulties and reduce usage under normal settings. A sensitive and consistent UPLC procedure was built to quantify NTG in the presence of another drug, metformin, with the help of Phenomenox C18 column (50×2.1 mm, 3.5 μm) using ammonium formate buffer of pH-3 and acetonitrile (75: 25, v/v) as mobile phase, eluted within 3 minutes at 260 nm. The linearity range of the reported method was in the range of 7.5–45 μg/mL with LOD and LOQ values of 0.09 and 0.3 μg/mL, respectively. The precision values were within the acceptable limit regarding % RSD between 0.45 and 0.58%.42
Whereas Waters Acquity UPLC SB C18 followed a slightly higher flow rate and column temperature of 30ºC to elute NTG at 1.71 min with a broader linearity range of 15–90 µg/mL.43 The method’s novelty was verifying the specificity test by forced degradation study. The UPLC method can quantify RPG in pharmaceutical formulations with precise and validated procedures within 2.1 minutes and reveals exceptional performance in terms of sensitivity and speed, which could be used as a routine RPG analysis. The method is stability-indicating for RPG assay in pure and formulations. Also, the method’s degradation study reveals it is precarious in acidic and basic forms.44
The product’s quality, as assessed by UPLC, would be better with less time. Still, the problem is related to column life because it requires high pressure, possibly damaging the column competence. Innovative and selective approaches were advanced to establish whether drugs with commercial formulations were single- or combined-dosage form
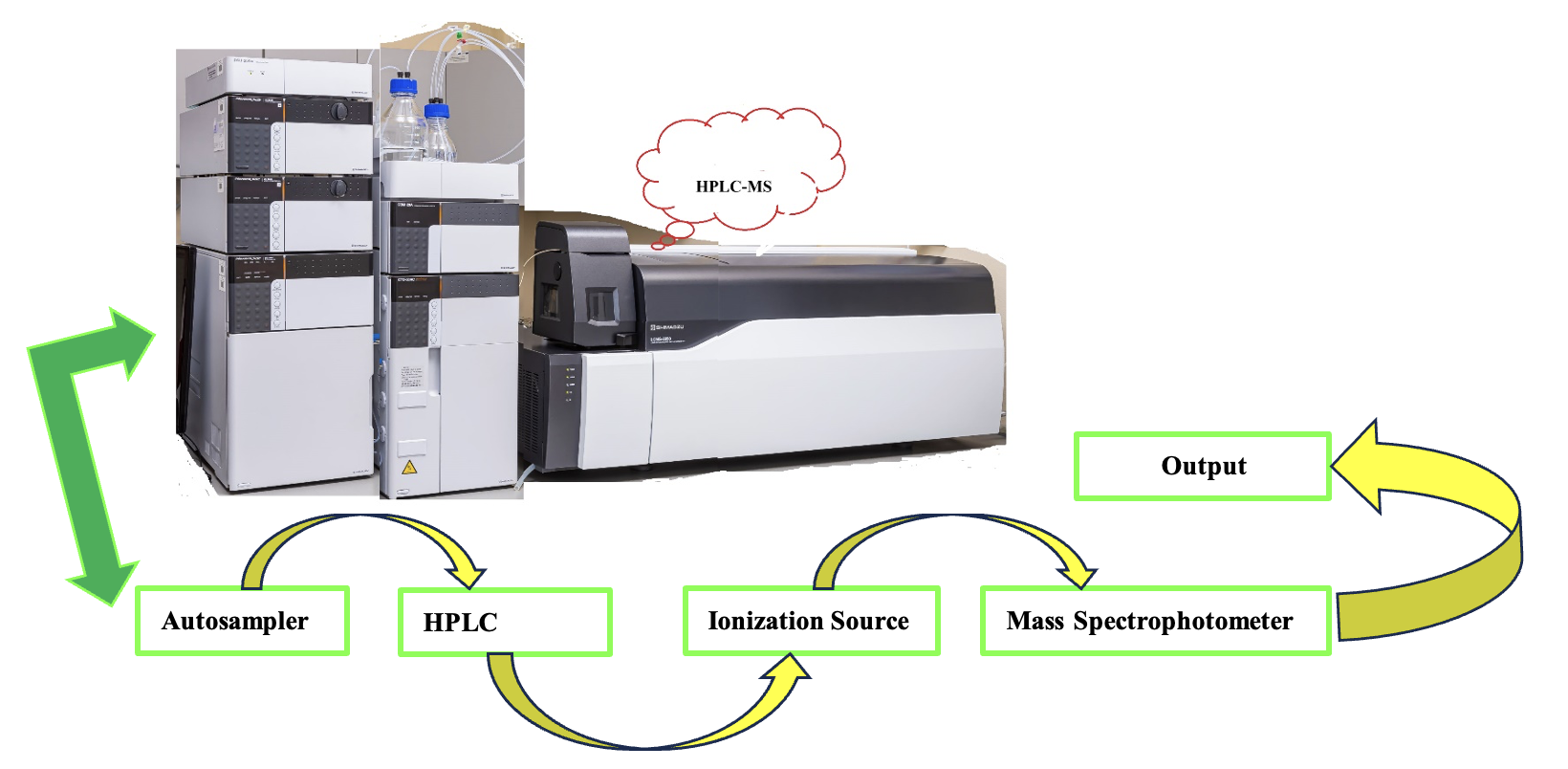
2.5. HPLC AND UPLC COMBINED MASS SPECTROPHOTOMETRY
LC is liquid chromatography combined with mass spectrometry (MS), either HPLC–MS (Scheme 5) or UPLC–MS (Scheme 6). The chromatography method’s separation ability and mass spectrophotometry identification facility are combined. MS is a perfect technique that can confirm a compound’s structure using its mass spectrum, and no standard is needed. Whereas LC-MS is a hyphenated practice, its fragmentation pattern could provide an idea of the structure of the compound with high selectivity and sensitivity.45 Also, the isotope peaks show the availability of oxygen, bromine and chlorine peaks.


These techniques are rapid and reliable for determining NTG and RPG as single or combined formulations with other drug substances or in the presence of their active metabolites. The LCMS determination extensively included lower amounts of biological samples46 to quantify NTG and RPG (Table 3). Additionally, fabric phase sorptive extraction (FPSE) enhanced the extraction process with a versatile and convenient practice that ensures lower LOQs, streamlining the method to a significant level.
The NTG’s degradation products were confirmed with LCMS and characterized as 2-amino-3-phenylpropanoic acid, 2-(cyclohexanecarboxamido)-3-phenylpropanoic acid and 4-isopropyl-N-phenethylcyclohexanecarboxamide. They can be done in acid, base, hydrolysis, and oxidative conditions.47 The technique was also enhanced by applying a C18 gold column, reducing its runtime and boosting the method’s ability for excellent sample throughput. The hydrophilic interaction liquid chromatography combined mass spectrophotometry-based assay was developed and validated for 13 antihyperglycaemic drugs, including NTG and RPG,48 in human urine samples (Figure 6). The HILIC instrumentation applied high-quality solvents as a mobile, which enhanced the assay method’s sensitivity and claimed to be the first simultaneous quantitative determination of 13 oral antihyperglycaemic drugs. The rapid method confirmed that no procedure was reported in any biological matrix for simultaneous quantification of NTG in a combined form with metformin.49 The kinetic parameters were determined in solutions, and nine degradation products of RPG were identified50 that could be beneficial for further findings to count them as new related substances (RS) in the pharmacopoeial profile (Figure 7). The UPLC/MS method was quantified with a broader analysis range of 500–4000 ng/mL with a 0.6 mL/min flow rate in plasma samples,51 including a fast and reproducible solution preparation technique to implement a simple protein precipitation method to study the bioequivalence, pharmacokinetic, therapeutic monitoring and bioavailability studies NTG, an antidiabetic drug agent. However, the parallel artificial liquid membrane extraction technique could be combined with UPLC electrospray ionization tandem mass spectrometry to estimate therapeutic RPG’s drug monitoring in diabetic patients efficiently.52 The developed PALME technique offered a straightforward sample preparation protocol for direct extraction from human plasma. The Analytical Green Calculator (AGREE) tool scrutinised PALME’s welfare and ecological impact, and the outcome was an eco-friendly assessment. All the results of the reported methods highlighted the advantage of the developed method in quantitating NTG and RPG from different sources in terms of sample throughput, reagents/solvent consumption, % recovery, selectivity and sensitivity.


3. METHOD DEVELOPMENT, ADVANTAGES AND LIMITATIONS OF THE PROPOSED TECHNIQUES
The drug development process begins with innovating a drug molecule with medicinal value in fighting, examining and curing diseases. It is necessary to characterize the synthesized molecule to initiate preliminary safety and therapeutic effectiveness by analyzing them, which are required to discover drug applicants for further investigations. Pharmaceutical development evidence requires scientific justification for formulation progress and a correct explanation of the dosage form. Regulatory assistance specifies limited information on the data constraints correlated with pharmaceutical development.
However, additional, comprehensive data is accessible for the toxicological estimation of excipients, which are a significant portion in the solid dosage forms, which assist as diluents to permit the formulation with correctly sized coverings from unwanted organoleptic abilities of the drug substance. The most common excipients, such as lactose, sucrose, cellulose, magnesium stearate, talc, calcium phosphate, starch, etc, are used in pharmaceutical formulations. In specific cases, excipients do not intermingle chemically but could help drug molecule degradation. It is also crucial to understand their chemical stability, impurity identification and quantification, which are beyond the recognised threshold vital to assess the impurities toxicity profiles to discriminate these from its active pharmaceutical ingredients (API) and pharmaceutical formulations.
Spectrophotometry is the numerical measurement of a substance’s reflection or transmission assets against wavelength, requiring minimal time and labour consumption. Due to its excellent precision, it enhanced its application in pharmaceuticals. However, the homogeneous solution necessary with optical density must not be low or high. Some components can dissolve only in nonpolar solvents and cannot be used in plastic cuvettes. Also, many components with close characteristic wavelengths in a sample could possibly obstruct each other, and pH needs to be verified cautiously since coloured components are pH-dependent.
Meanwhile, the advanced liquid chromatography technique, which is liable to HPLC, likely helps separate molecules in a complex chemical and biological matrix. The specificity, accuracy and precision results are excellent. However, it also stated that these parameters are only attainable when suitability tests are performed with a wide range. In HPLC, the detection is crucial to ensure all the components are detected. HPLC-UV detectors are widely used and can monitor a range of wavelengths simultaneously. Over a certain period, researchers mostly used UV absorbance with the reversed-phase mode whenever suitable. This offered reliability, sensitivity, repeatability, and analysis time but was restricted by column price, solvents, and column packing materials, ultimately affecting long-term reproducibility. HPTLC is an advanced form of thin-layer chromatography (TLC). Several amendments were made to the primary method of TLC to automate the discrete steps to achieve the enhanced resolution and limit of detection. However, it required a large volume of expensive organics, stringent operation conditions, mainly dust-free environment and temperature control and highly skilled and trained people to run the system. It is also limited to plate length and is indirectly restricted to a specific length for component separation. The developed UPLC promotes advancement in three main areas: sensitivity, resolution, and speed. This technique could work with less than 2 µm fine particles.
Consequently, it reduced the column length and ultimately decreased solvent consumption and run time. However, increased column pressure minimises column life and is necessary for more maintenance. The separation technique’s coupling with another leads to the development of hyphenated techniques like LC-MS and UPLC-MS. The LC-MS method is rapid analysis. Quantifying several analytes requires a short time and quickly determining parent drug and their metabolites. However, highly trained and skilled people must apply high-cost reagents, sample pretreatment before each analysis, and complex sample preparation. The UPLC-MS technique is robust, proficient in bioequivalence study, and straightforward for biological fluids with lower LOD and LOQ values. It needs sample derivatization and multi-reaction monitoring with high-cost equipment.
These drugs are noteworthy and are one of the fastest-growing products in the pharmaceutical industry due to their increasing demand. The spectrophotometers are simple to supervise and available everywhere due to their low cost. The biological fluids and trace analysis can be used for susceptible instruments like HPLC, HPTLC, and UPLC combined with the mass spectrophotometer. Every technique has advantages and limitations (Table 4), which is also essential to selecting the correct methods for particular samples, depending on the capacity and capability, but never compromising the valuable data directly related to the efficacy and safety of drug products and human health. The innovations of nanotechnology, artificial intelligence, and green analytical chemistry drastically improve the precision and productivity of pharmaceutical analysis to safeguard that drugs execute their expected intent. Various instrumental methods have been built over time for drug evaluation. Therefore, analytical instrumentation and techniques are necessary for identifying and quantifying these drug impurities. Pharmaceutical manufacturing is constantly changing, and there is a considerable attempt to translate new regulatory requirements into practice by implementing new technologies.
4. CONCLUSION
The present review discussed the summary of the reported method as per the literature for estimating RPG and NTG, not only in bulk and pharmaceutical formulations but also in biological matrices. Analytical techniques such as UV-Vis, HPLC, HPTLC, UPLC, HPLC/MS and UPLC/MS were employed to determine drugs for quality control samples. The main objective is to provide academicians, researchers, and scientists with appropriate information about glitinide agents. After studying each technique’s capabilities and limitations, people may understand the drug’s behaviour and easily enhance new technology that could benefit human health and provide a better, greener methodology. These drugs are critical due to their mounting demands in our daily lives, which is the crucial reason for manufacturing more products in the pharmaceutical industry. HPLC and UV-Vis technology are low-cost, easy to handle, and available, boosting their application for pure and pharmaceutical dosage forms. The biological fluids and trace analysis would go for susceptible instruments combined with a mass spectrophotometer. Therefore, the present review discusses various analytical methods and compares efficacy when applied to NTG and RPG. Also, ideally, it provides insights into which strategies are most effective under specific conditions for determination and quantification in API, pharmaceutical tablets, and biological fluids.
FUNDING
This research did not obtain any financial grant to complete the work.